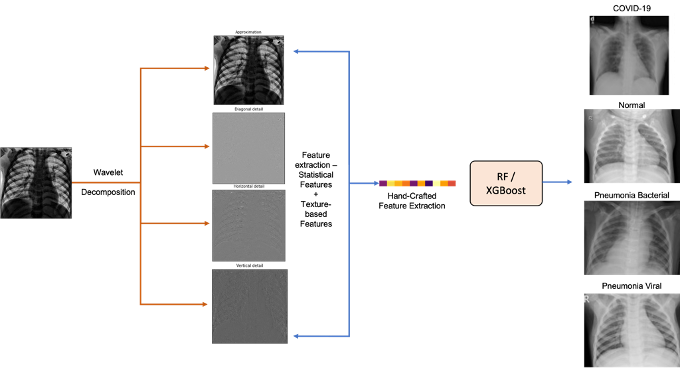
Wavelet Transform and Machine Learning-Driven Multi-Class Classification of Chest X-Ray Images for COVID-19 Diagnosis
Chest radiography is a fast, low cost, and widely available first line test for suspected pneumonia and other respiratory infections, which makes automation clinically valuable where CT and subspecialty radiologists are limited. Yet adoption of automated CXR analysis using machine learning has been slowed by the memory and compute demands of large deep convolutional networks, their training time, and the need for GPU infrastructure, which limit deployment at the edge and in resource constrained settings. In acute care, separating COVID-19, bacterial pneumonia, and viral pneumonia is necessary for isolation, preventive care, so the classification is valuable and directly actionable by hospitals and clinics. We present a lightweight pipeline for multiclass CXR classification that applies a single level discrete wavelet transform and computes 11 statistical and texture descriptors per subband, producing a 44 dimensional feature vector for tabular classifiers. We evaluate CXRs stratified across four classes (healthy, COVID-19, bacterial pneumonia, viral pneumonia). Random Forest and XGBoost reach 96.83% and 97.76% test accuracy, respectively, outperforming transfer learned DCNN baselines on the same data. Ablations show that features pooled across subbands outperform any single subband and that texture descriptors including entropy, contrast, homogeneity, dissimilarity, and correlation carry most of the signal. Among wavelet bases, single level Bior6.8 performs best, while deeper decompositions reduce accuracy at this data scale. Because computation is limited to a grayscale wavelet transform and tabular learning, the method runs on commodity CPUs, has a small memory footprint, and is simple to reproduce, with strong per class performance for COVID-19, bacterial pneumonia, and viral pneu- monia that supports further treatment.